Is ISO Certification Worth It? A Cost-Benefit Guide for Businesses
ISO certification is worth it when it helps a business win contracts, meet customer requirements,…

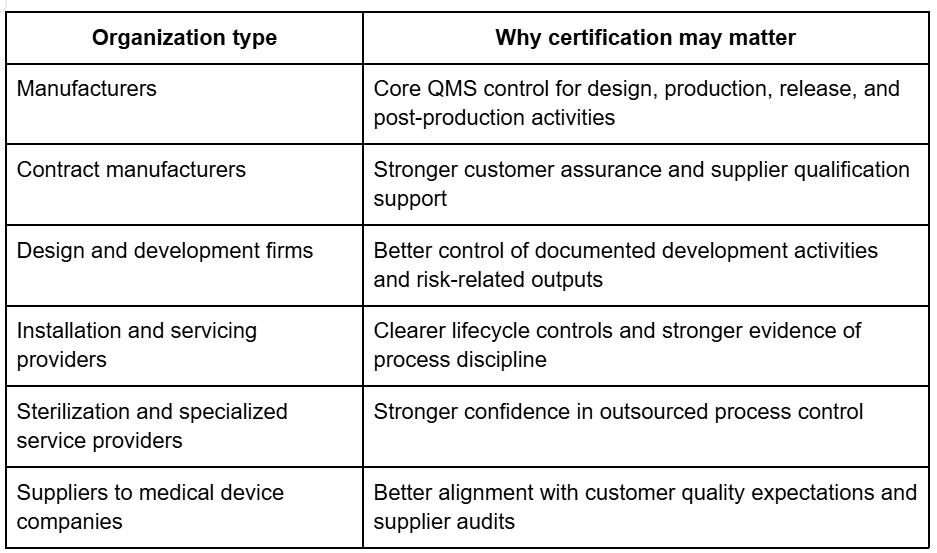
ISO 13485 certification is relevant across a wider range of medical device roles than many companies expect. It is not limited to large manufacturers.
It is commonly pursued by:
A simple way to judge fit is this: if your work can affect device quality, safety, conformity, or controlled lifecycle activities, ISO 13485 is probably relevant.
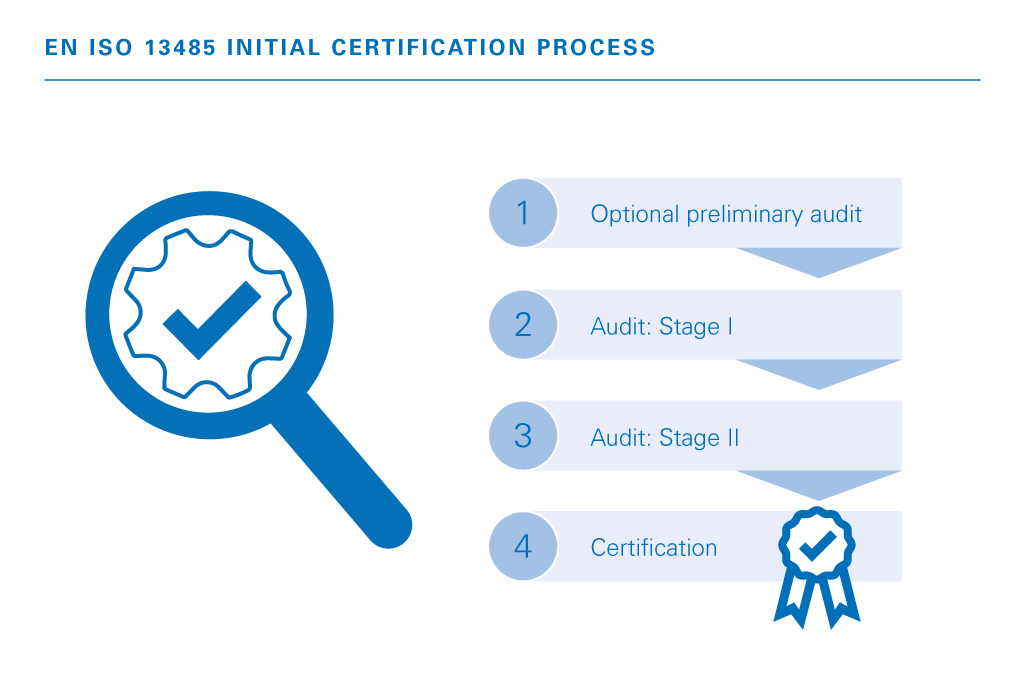
The certification path is straightforward when the groundwork is real.
A rushed certification timeline usually creates more cost, more rework, and worse audit outcomes. A realistic timeline usually leads to better audit outcomes and lower overall cost.
AGS can review your scope, current QMS maturity, supplier-control model, and likely audit friction points before you commit to a certification timeline.
Book an ISO 13485 readiness review or request a gap assessment.










If your organization is evaluating ISO 13485 certification seriously, the next step should be a structured scoping conversation, not a generic price request.
A useful first discussion should cover:
AGS can help you sort that out before time and money get wasted on the wrong sequence.
Talk with AGS about your device scope, current QMS, and audit readiness.







