Is ISO Certification Worth It? A Cost-Benefit Guide for Businesses
ISO certification is worth it when it helps a business win contracts, meet customer requirements,…

ISO 13485 is the international quality management system standard for organizations involved in one or more stages of the medical device life cycle. ISO says it is intended for organizations involved in design, production, installation, servicing, and related services for medical devices. IMDRF uses the same life-cycle framing and makes clear that the standard applies across one or more stages of the medical device life cycle, not only to large finished-device manufacturers.
The unique part is this: ISO 13485 is written for regulatory purposes. That is what makes it different from generic quality content. ISO’s own title says so. ISO’s guidance on the 2016 revision also says the standard puts greater emphasis on risk management and risk-based decision-making outside product realization.
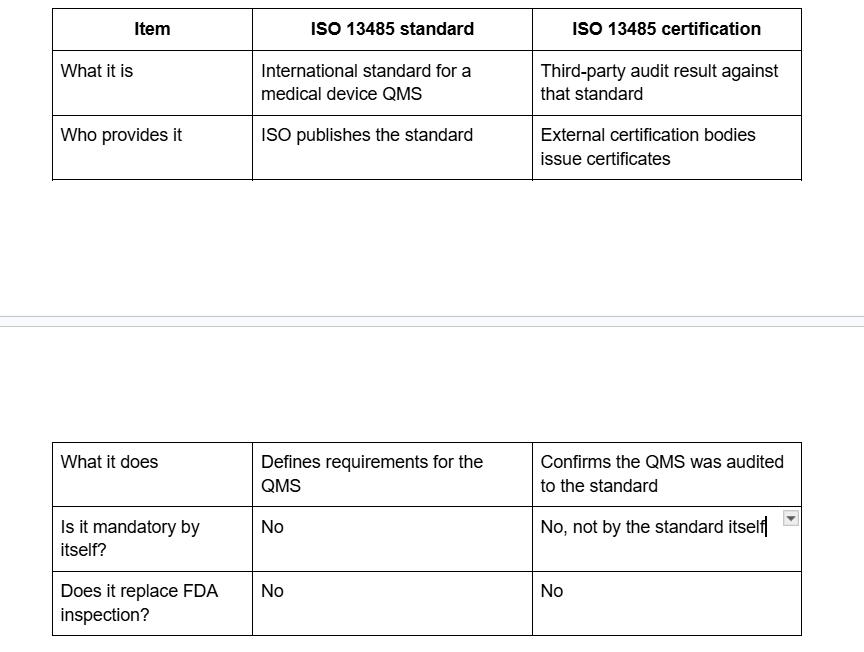
ISO says management system certification is not required by the standard itself, and ISO says it does not perform certification. FDA says a certificate of conformance to ISO 13485 does not exempt a manufacturer from FDA inspection.
ISO 13485 certification proves that a certification body audited the organization’s quality management system against ISO 13485 requirements. ISO 13485 certification does not prove that every product is safe in every case, and it does not create automatic regulatory approval. FDA says plainly that an ISO 13485 certificate does not replace FDA inspection and that FDA does not issue those certificates.
Organizations that work in one or more stages of the medical device life cycle are the main fit for ISO 13485. ISO says that includes design, production, installation, servicing, and related services. IMDRF extends that same logic across one or more stages of the medical device life cycle.
The root fit question is not size first. It is role first. If the organization affects product quality, regulatory evidence, traceability, servicing, or supplier control in the medical device chain, ISO 13485 becomes relevant fast.
What types of companies use ISO 13485?
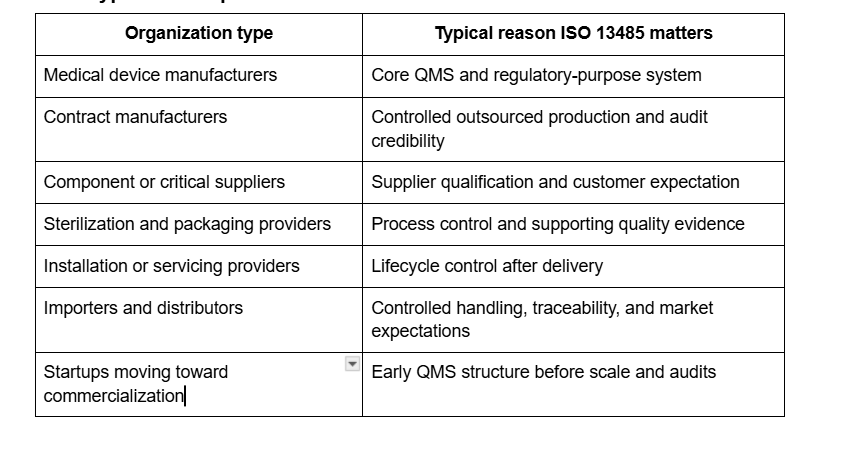
ISO’s scope language supports this breadth, and ANAB’s accreditation language confirms that certification bodies audit organizations using ISO 13485 in that medical device QMS context.
Yes. ISO says ISO 13485 can benefit suppliers and external parties that provide product or quality-management-system-related services to medical device organizations. That means component suppliers, contract service providers, sterilization vendors, calibration providers, and other support organizations can pursue certification when customer requirements or supplier-approval expectations make it commercially valuable.

















